Fragment picking documentation
Metadata
Author: Dominik Gront (dgront@chem.uw.edu.pl)
Last edited 4/22/11. Corresponding PI Dominik Gront (dgront@chem.uw.edu.pl).
Code and Demo
Application source code:
rosetta/rosetta_source/src/apps/public/fragment_picker.cc-
Try-me examples The examples are located in rosetta/rosetta_demos/fragment_picker directory:
- BestFragmentsProtocol : basic example demonstrates how to select fragments that maximize a given score function
- QuotaProtocol : quota protocol demo shows how to pick fragments for an ab-initio protein prediction
- Quota_with_restraints : demonstrates how to incorporate distance restraints to a fragment picking run
To run picker, type the following in a commandline:
[path to executable]/picker.[platform|linux/mac][compile|gcc/ixx]release –database [path to database] @options
References
The algorithm and the code components have been described in:
- Gront D, Kulp DW, Vernon RM, Strauss CEM and Baker D, "Generalized fragment picking in Rosetta: design, protocols and applications", submitted to PLoS ONE
Purpose
Pick fragment sets for Rosetta protein structure modeling
Algorithm
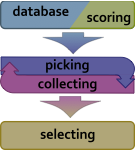 Figure 1: A general scheme of the fragment picking workflow Figure 1: A general scheme of the fragment picking workflow |
Detail of the algorithm are described in Gront D. et al paper. In brief, the program reads a database file (nicknamed vall ), input query sequence or sequence profile and other files and produces fragment files for modeling with Rosetta. The picking process consists of three stages: preparation (reading input files, etc), actual fragment picking when the candidates are pushed into a collector, and a selection when the final fragment set is prepared based on the collected candidates. |
|---|
Modes
The picker provides three fragment picking protocols:
- Quota - substitutes the old
nnmakeprogram; selects fragments suitable for de-novo protein structure prediction - Best fragments - selects fragments by maximizing score function; the protocol works well if available data (e.g. local backbone NOE, Chemical Shifts, restraints) are satisfactory to define the optimal solution
- Keep all - may be used to enumerate all the fragments that satisfy some criteria. In general the protocol consumes a lot of memory and should be used only for very particular applications.
Input Files
There are many possible input files, depending on the picking protocol and scoring function. The most commonly used are:
| file type | description | where does it come from | who uses it |
|---|---|---|---|
| vall | protein structures database, your fragments come from there. | should be in Rosetta SVN repository. A copy of this file can be also downloaded from: http://www.bioshell.pl/rosetta-related/vall.apr24.2008.extended.gz | mandatory file |
| .wghts | defines scoring system for fragment selection | edit one of the examples provided below | mandatory file |
| .fasta | amino acid sequence | you must already have it... | mandatory file unless .chk is given |
| .chk | sequence profile created with PSI-Blast with further modifications (pseudocounts added) | make_fragments.pl script | any sequence profile - based score, e.g. ProfileScoreL1; mandatory file unless .fasta is given |
| .ss2 | secondary structure prediction in PsiPred format | The easiest way is to run make_fragments.pl script. You may also try to run a secondary prediction software on your own and then convert the resulst to the proper format. A script convert_ss_predictions.py can turn TALOS, Juffo, Porter and SAM into ss2. | SecondarySimilarity or SecondaryIdentity scores |
| .cst | distance (or dihedral) constraints | Convert your data (distances or torsion angle values) into the proper format. | AtomPairConstraintsScore or DihedralConstraintsScore scores |
| .tab | chemical shifts in TALOS format | NMR experiment; examples can be downloaded from BMRB database | CSScore (CS-Rosetta protocol) |
| .pdb | reference structure in PDB format | used for fragments' quality assessment |
Note, that some of these files are produced by external programs:
| input data | program | notes |
|---|---|---|
| sequence profile | PsiBlast (the old version, not the C++ one!) | Raw PsiBlast checkpoint is stored in a binary format. The file is processed by make_fragments.pl script that adds pseudocounts to empty rows in the profile and saves it in a flat text format |
| secondary structure prediction | PsiPred, Jufo, SAM, Porter | All these programs require PsiBlast to be installed.fragment_picker reads input secondary structure predicitons only in PsiPred's SS2 format. The ss_pred_converter.py script may be used to convert from other file formats |
Weight file for fragment picking
A weight file has at least four columns, which provide: score name, its priority, weight and the maximum allowed value. If for a certain candidate a given score returned value higher than the maximum allowed, the fragment candidate is no longer considered and any further score won't be evaluated. The scores are evaluated according to the decreasing priority rather than the order how they are listed in a weight file. To be sure that all scores are evaluated for each fragment, put '-' (dash) character as the max_allowed score value.
Weight value 0.0 has a special meaning: such scores are evaluated only for the selected fragments, at the end of a program where output files are written. This allows reduce the time spent on fragments descriptive statistics evaluation, such as crmsd or Gunn cost.
Typical weight values are given below:
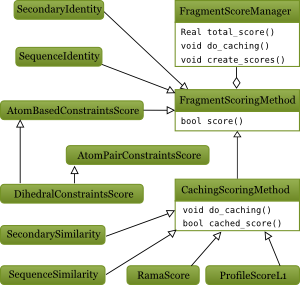 Figure 2: UML diagram of the core fragment scoring classes
Figure 2: UML diagram of the core fragment scoring classes
For ab-initio prediction (quota protocol):
# score name priority wght max_allowed extras
SecondarySimilarity 350 1.0 - psipred
SecondarySimilarity 300 1.0 - sam
SecondarySimilarity 250 1.0 - porter
RamaScore 150 2.0 - psipred
RamaScore 150 2.0 - porter
RamaScore 150 2.0 - sam
ProfileScoreL1 200 2.0 -
PhiPsiSquareWell 100 0.0 -
FragmentCrmsd 30 0.0 -
CS-Rosetta style fragment picking:
# score name priority wght max_allowed extras
CSScore 375 3.0 -
RamaScore 400 2.0 - talos
SecondarySimilarity 350 3.0 - talos
ProfileScoreL1 200 1.0 -
PhiPsiSquareWell 100 0.0 -
FragmentCrmsd 30 0.0 -
GunnCostScore 20 0.0 -
```
Everything that starts at the fifth column goes to a score term maker as additional parameters. The most important application is to provide secondary structure prediction name for quota protocol.
Options
=======
Protocol-Specific Options
-------------------------
|option|description|example|
|------|-----------|-------|
|in:file:native|Native PDB filename|2gb1.pdb|
|in:file:vall|vall database for fragment picking|vall.dat.apr24.combo.aug09|
|in:file:s|Name(s) of single PDB file(s) to process|2gb1.pdb|
|in:file:xyz|Input coordinates in a raw XYZ format (three columns)|2gb1.xyz|
|in:file:fasta|Fasta-formatted sequence file|2gb1.fasta|
|in:file:pssm|NCBI BLAST formatted position-specific scoring matrix|2gb1.pssm|
|in:file:checkpoint|Sequence profile (binary file format) prepared by NCBI BLAST|2gb1.checkpoint|
|in:file:talos\_phi\_psi|File that provides Phi-Psi angles in Talos+ format|2gb1.tab|
|in:file:torsion\_bin\_probs|File describing probabilities over torsion bins A,B,E,G,O|2gb1.bin\_probs|
|in:path:database|Database file input search paths. If the database is not found the ROSETTA3\_DB environment variable is tried.||
|frags:scoring:config|scoring scheme used for picking fragments|quota\_scores.wghts|
|frags:scoring:profile\_score|scoring scheme used for profile-profile comparison|L1|
|frags:ss\_pred|provides one or more files with secondary structure prediction (PsiPred SS2 format) , to be used by secondary structure scoring and quota selector. Each file name must be followed by a string ID.|2gb1.psipred\_ss2 psipred 2gb1.jufo\_ss2 jufo 2gb1.sam\_ss2 sam|
|frags:n\_frags|number of fragments per position|200|
|frags:n\_candidates|number of fragment candidates per position; the final fragments will be selected from them|1000|
|frags:frag\_sizes|sizes of fragments to pick from the vall|3 9|
|frags:write\_ca\_coordinates|Fragment picker will store CA Cartesian coordinates in output fragment files. By default only torsion coordinates are stored.||
|frags:allowed\_pdb|provides a text file with allowed PDB chains (five characters per entry, e.g.'4mbA'). Only these PDB chains from Vall will be used to pick fragments|templates.pdb\_ids|
|frags:denied\_pdb|provides a text file with denied PDB chains (five characters per entry, e.g.'4mbA'). This way close homologs may be excluded from fragment picking.|homologs\_vall|
|frags:describe\_fragments|Writes scores for all fragments into a file|frags.fsc|
|frags:keep\_all\_protocol|makes the picker use keep-all protocol to select fragments. The default is bounded protocol||
|frags:bounded\_protocol|makes the picker use bounded protocol to select fragments. This is the default behavior||
|frags:quota\_protocol|quota protocol implies the use of a QuotaCollector and a QuotaSelelctor, no matter what user set up by other flags.||
|frags:picking:selecting\_rule|the way how fragments are selected from candidates, e.g. QuotaSelector of BestTotalScoreSelector|BestTotalScoreSelector|
|frags:picking:quota\_config\_file|provides a configuration file for quota selector|quota.conf|
|frags:picking:query\_pos|provide sequence position for which fragments will be picked. By default fragments are picked for the whole query sequence|21 22 23 24 25 26 27 28 29|
|constraints:cst\_file|constraints filename(s) for centoroid. When multiple files are given a *random* one will be picked.|2gb1-noe.cst|
|out:file:frag\_prefix|Prefix for fragment file output|aa|
The fragment picker components and concepts
===========================================
In brief, the picker process vall database one chunk after another. For each chunk it takes all possible fragment candidates, scores them and stores inside collectors. When all vall chunks are processed, the collectors' content is passed to a selector which selects the final fragments. These are saved into file(s). All parts of this machinery are briefly described below.
Fragment candidate
------------------
... is a fragment-to-be, if it survive the collection and selection stages.
Fragment collector
------------------
The collector collects fragments along with their scores; all the colectors are build on utility::vector1d7f0aea0dac652499a39ec092bda52aacd796fe6lt;d7f0aea0dac652499a39ec092bda52aacd796fe6gt;. Unfortunately there are more than 2M possible fragment candidates. To keep them all one would need about ... per each residue in a query sequence. Therefore a collector may keep only a small fraction of all candidates. BoundedColelctor keeps Ncand best candidates per each position in a query sequence, where "best" is defined by a comparator object that is used to sort the container.
Fragment selector
-----------------
Fragment selection rule takes all fragment candidates and selects the final Nfrags fragments.
Cacheable fragment score
------------------------
Caching is a way to speed up fragment scoring by recycling per-residue score values. Caching score function must implemant do\_caching() method which evaluates a full matrix of pairwise residue-vs-residue scores. For instance ProfileL1Score compares any column from query profile with any profile column from a chunk. When it comes to compute a score of a fragment of length nf that start at qi in query and at ci in chunk, a simple sum over a stripe qi-d7f0aea0dac652499a39ec092bda52aacd796fe6gt;qi+nf; ni -d7f0aea0dac652499a39ec092bda52aacd796fe6gt; ni+nf is computed. Moreover, to evaluate a score for the very next fragment (i.e. the one staring at (qi+1,ni+1)), one can just has to subtract one and add one per-residue score.
Obviously caching doesn't work when a score cannot be decomposed into per-residue components, e.g. FragmentCrmsd or RDCScore. In some cases caching is actually slower than just computing the score without caching, e.g. SequenceIdentity
Quota system
------------
In general the purpose for quota is to keep the diversity within fragments. If for example a given position in a query sequence has been predicted to be helical with 70% chance and loop with 30%, "select best" protocol will pick only helical fragments for this position, because they will be favored by the ` SecondarySimilarity ` scoring term. To the contrary, quota protocol will pick 30% (best scoring) loop fragments and 70% best scoring helices. The situation is more complicated by the fact that 3 secondary structure predictors are used. This makes in total 9 different categories of fragments (referred further as quota pools) collected and scored separately. Once final fragments are selected (separately for each quota pool), they are merged into a single set.
Quota protocol uses quota specific collectors and selectors. Scoring scheme is also altered.
Quota pools
-----------
In quota protocol there are several fragment categories (pools), that are kept separated from each other. They are collected, scored and selected separately. By default there are 3 secondary structure predictions used for fragment picking: PsiPred, SAM and Porter. The fragment candidates are also split by the secondary structure class (H, E or L) which makes 9 quota pools in total. The size of each pool is controlled by quota allowance and secondary structure probability.
From the implementation's point of view, a quota pool is a BoundedCollector whose size is based on quota allowance, sorted by slightly modified quota score. Note, that quota pools, similarly to fragment collectors, are position specific, so for a 100aa query sequence there are about 900 quota pools.
Quota.def file
--------------
pool_id pool_name fraction
1 psipred 0.6 2 porter 0.2 3 sam 0.2
Quota allowance
---------------
is defined for each predictor by a Quota.def file. Default allocations are: PsiPred - 0.6 SAM - 0.2 Porter - 0.2 Final allowance for a quota pool is a product of predictor share and secondary structure probability. For example, if PsiPred predicted that a certain position is helical
Quota score - pool identification
---------------------------------
As it has been mentioned in [Quota score](#Quota-score) section, some scores are switched on and off for different pools. To have it working properly, the two config files: [Weight file for fragment picking](#Weight-file-for-fragment-picking) and [Quota.def file](#Quota.def-file) must contain matching string identifiers. Although the above examples use the predictors' names (psipred, porter and sam) for this purpose, one can use any arbitrary strings. The only limitation is that the three :
- secondary prediction name given in a weight file
- quota pool name, given in Quota.def file
- econdary prediction name assigned to a RamaScore or SecondarySimilarity score must match.
Quota score
-----------
The only difference between the fragment total score and fragment quota score is in the use of proper secondary-structure variant of some scores. Currently this only implies to RamaScore and SecondarySimilarity score. So for example, a quota pools created from a prediction named "psipred" use **only** SecondarySimilarity score named "psipred".
Tips
====
- multiple fragment sizes:
- say ` -frags:frag_sizes 3 4 5 ` to pick 3-mers, 4-mers and 5-mers
- picking fragments for a predefined region in the query sequence:
- say ` -frags:picking:query_pos 21 22 23 24 25 26 27 28 29 ` to pick fragments at the selected positions
- test for homologues contamination: In order to obtain objective results from ab-initio protein structure prediction bechmarks, fragment sets should not contain pieces of homologous proteins. The PDB codes of unwanted protein chains should be listed in a file and provided to the picker with -frags:denied\_pdb flag. This however does not quarantee the results are free from homologous fragments. It is recommended to check which PDB entries contribute to a fragment, e.g. by the following bash command:
cat aa2gb1.9mers | awk '{print $1}' | uniq | grep -v 'position' | grep '.' | sort | uniq -c | sort -n
Homologues structures introduces significantly more fragments than unrelated proteins. One should manually examine the most popular hits, possibly add them to the list of denied PDB ids and run the fragment picker once again.
Expected Outputs
================
There are two kinds of output files:
fragment file
-------------
Output fragments are written in Rosetta++ format.
fragment score file
-------------------
Fragment scores are stored in a flat tabulated format, one score file for each fragment size. All columns from a single line describe a single fragment and provide:
- its first residue index in a query sequence
- its first residue index in the source vall chunk
- PDB id of the source vall chunk
- chain id of the source vall chunk
- secondary structure type for the middle residue of the fragment
- all score values for the whole fragment, ordered by their priority (descending)
- quota-specific total score for this fragment (only in the case of quota protocol)
- total (weighted) score for this fragment
- name of the quota pool who contributed this frgament (only in the case of quota protocol)
- unique integer ID of this fragment; this is defined as a line number from the vall file where the data for the first residue for this fragment is stored. If user provided more than one vall file, continuous numbering is used.
Post Processing
===============
Fragment may be directly used by Rosetta 2.x and 3.x. Fragment score file may be useful for debuging, check for quota levels, fragment quality assessment, etc.
New things since last release
=============================
This is the first public release